商品详情
返回产品目录
商品包装及说明书因厂家更换频繁,如有不符以实物为主
洛那法尼布片
国际零售参考价:¥**/盒
-
温馨提醒:本说明书仅供参考,最新的说明书详见药品附带的说明书。
1 适应症和用法
ZOKINVY适用于 12 个月及以上、体表面积 (BSA) 为 0.39 m 2及以上的患者:
- 降低哈钦森-吉尔福德早衰综合征 (HGPS) 的死亡风险
- 用于治疗加工缺陷的早老性椎板病:
- 具有 progerin 样蛋白积累的杂合LMNA突变
- 纯合或复合杂合ZMPSTE24突变
使用限制
ZOKINVY不适用于其他早老症综合征或处理熟练的早老症椎板病。根据其作用机制,预计ZOKINVY不会对这些人群有效。
2 剂量和给药方法
2.1 推荐用量
- ZOKINVY对 BSA 为 0.39 m 2及以上患者的起始剂量为 115 mg/m 2每天两次与早晚饭(见表 1)以降低胃肠道不良反应的风险[见不良反应(6.1)] . ZOKINVY的适当剂量强度不适用于 BSA 小于 0.39 m 2 的患者[见适应症和用法 ( 1 )]。
- 治疗 4 个月后,将剂量增加至 150 mg/m 2,每天2次,早晚各一次(见表 2)。
- 将所有每日总剂量四舍五入到最接近的 25 毫克增量(见表 1和表 2)。
- 如果错过剂量,请在下一次预定剂量前最多 8 小时随食物一起服用。如果距下一次预定剂量还有不到 8 小时,则跳过错过的剂量,并在下一次预定剂量下继续服用ZOKINVY。
表 1提供了基于 BSA 的剂量建议,用于每天两次115 mg/m 2的起始剂量。

表 2提供了每日两次150 mg/m 2剂量的基于 BSA 的剂量建议。

2.2 胃肠道不良反应的剂量调整
对于已将ZOKINVY的剂量增加至 150 mg/m 2每天两次并且正在经历反复呕吐和/或腹泻导致脱水或体重减轻的患者,ZOKINVY 的剂量可以减少至 115 mg/m 2的起始剂量每天两次(见表 1)。确保ZOKINVY与早晚餐和足量的水一起服用。
2.3 药物相互作用的剂量调整
CYP3A 抑制剂
如果ZOKINVY与弱 CYP3A 抑制剂的同时使用是不可避免的[见警告和注意事项 ( 5.1 ),药物相互作用 ( 7.1 )]:- 以每天两次115 mg/m 2的起始剂量减至或继续ZOKINVY(见表 1)。
- 在停止同时使用弱 CYP3A 抑制剂后 14 天恢复先前的ZOKINVY剂量。
2.4 咪达唑仑的暂时停用
在咪达唑仑给药前 10 至 14 天和给药后 2 天暂时停用ZOKINVY [见禁忌症 ( 4 ),药物相互作用 ( 7.2 )]。
2.5 制备和给药说明
辖ZOKINVY口服早上和晚上的饭菜。
能够吞咽胶囊的患者
- 辖ZOKINVY胶囊全用足量水。不要咀嚼胶囊。
患者无法吞咽胶囊
- ZOKINVY胶囊的全部内容物可以与 Ora Blend SF ®或 Ora-Plus ®混合,或者,对于无法接触或耐受 Ora Blend SF 或 Ora-Plus 的患者,ZOKINVY胶囊的内容物可以与橙汁或苹果酱混合(请参阅下面的准备说明)。
- 不要与含有葡萄柚或塞维利亚橙子的果汁混合[见禁忌症 ( 4 ),药物相互作用 ( 7.1 )]。
- 每种剂量的混合物必须新鲜制备,并在混合后约 10 分钟内服用。
Ora Blend SF、Ora-Plus 或橙汁中剂量的制备
- 对于每个胶囊,将胶囊的内容物倒入装有 5 mL 至 10 mL 液体的容器中。
- 用勺子彻底混合。
- 消耗整个服务。
苹果酱中剂量的制备
- 对于每个胶囊,将胶囊的内容物倒入装有 1 茶匙至 2 茶匙苹果酱的容器中。
- 用勺子彻底混合。
- 消耗整个服务。
3 剂型和规格
胶囊:
- 50 mg,不透明黄色,黑色印有“LNF”和“50”
- 75 毫克,不透明的浅橙色,带有黑色印有“LNF”和“75”的字样
4 禁忌症
ZOKINVY禁用于服用以下药物的患者:
5 警告和注意事项
5.1 由于药物相互作用而降低疗效或不良反应的风险
ZOKINVY与其他药物的共同给药可能导致临床上显着的药物相互作用[见剂量和给药方法 ( 2.3 , 2.4 ), 禁忌症 ( 4 ), 药物相互作用 ( 7.1 , 7.2 )]。这些药物相互作用可能导致:
- ZOKINVY 的疗效降低
- ZOKINVY或合用药物的不良反应风险增加
有关预防或管理这些具有临床意义的药物相互作用的步骤,请参见表 4和表 5,包括剂量建议[参见药物相互作用 ( 7.1 , 7.2 )]。在ZOKINVY治疗之前和期间考虑药物相互作用的可能性;在ZOKINVY治疗期间审查伴随药物;并监测不良反应。
5.2 实验室异常
一些用ZOKINVY治疗的患者出现实验室异常[见不良反应 ( 6.1 )]。这些包括:
- 电解质异常 (43%),例如高钾血症、低钾血症、低钠血症或高钙血症
- 骨髓抑制 (35%),例如中性粒细胞绝对计数、白细胞计数、淋巴细胞、血红蛋白或血细胞比容减少
- 肝酶升高,如天冬氨酸转氨酶 (35%) 或丙氨酸转氨酶 (27%)
这些实验室异常在继续ZOKINVY 时通常会改善,但不可能排除ZOKINVY作为异常的原因。定期监测电解质、全血细胞计数和肝酶,并相应地处理异常情况。
5.3 肾毒性
Lonafarnib 在大鼠血浆药物暴露引起肾毒性大约等于用人剂量达到的[见非临床毒理学( 13.2 )]。在ZOKINVY治疗期间定期监测肾功能。
5.4 视网膜毒性
Lonafarnib 在与人类剂量达到的血浆药物暴露相似时在猴子中引起视杆依赖性、低光视力下降[见非临床毒理学 ( 13.2 )]。定期进行眼科评估,并在ZOKINVY治疗期间出现任何新的视觉变化时进行。
5.5 生育能力受损
Lonafarnib 在雌性大鼠中基于血浆药物暴露在人剂量的 1.2 倍时导致生育力受损[见非临床毒理学 ( 13.1 )]。
Lonafarnib 导致雄性大鼠生育力受损和睾丸毒性基于血浆药物暴露在人剂量的 1.5 倍[见非临床毒理学 ( 13.1 )],并且在猴子雄性生殖道中的毒性低于基于血浆药物的人剂量暴露[见非临床毒理学( 13.2 )]。
忠告雌性和雄性动物生育能力研究结果的生殖潜力,尚未充分评估对青春期发育的影响和ZOKINVY治疗对人类生育能力受损的可能性[见在特殊人群中使用( 8.3 )]。
5.6 胚胎-胎儿毒性
根据动物生殖研究的结果,孕妇服用 ZOKINVY会造成胚胎-胎儿伤害。在动物生殖研究中,妊娠大鼠在器官形成期间口服 lonafarnib 在血浆药物暴露约等于推荐的人用剂量时产生胚胎-胎儿毒性。在怀孕兔中,在器官形成期间口服 lonafarnib 会在低于人类暴露的暴露下产生骨骼畸形和变异。告知孕妇对胎儿的风险。忠告有生育潜力的女性在用ZOKINVY 治疗期间避免怀孕和使用适当有效的避孕措施[见特殊人群中的使用 ( 8.1 , 8.3))]。
6 不良反应
6.1 临床试验经验
因为临床试验是在广泛不同的条件下进行的,在一种药物的临床试验中观察到的不良反应率不能直接与另一种药物临床试验中的发生率进行比较,并且可能无法反映实践中观察到的发生率。
总共 84 名受试者接受了至少一剂ZOKINVY 的治疗,有或没有额外的治疗,其中 8 人以至少 115 mg/m 2的剂量每天两次治疗大于或等于 10 年。
ZOKINVY的安全性基于 128 患者年的治疗暴露(62 例 HGPS 患者和 1 例处理缺陷型早老性椎板病伴LMNA杂合突变)和两项 2 期开放标签单臂试验的汇总结果( n=63:来自研究 1 的 28 名患者和来自研究 2 的 35 名初治患者)。在研究 1 中,ZOKINVY治疗开始于 115 mg/m 2每天两次并在大约 4 个月后增加至 150 mg/m 2每天两次,总治疗持续时间为 24 至 30 个月。研究 2 中的初治患者接受ZOKINVY 150 mg/m 2每天两次,共长达 36 个月。在这两项研究中,ZOKINVY通过胶囊口服给药,或者将胶囊内容物与 Ora Blend SF 或 Ora-Plus 混合并作为悬浮液口服给药。
在这两项研究中,共有 63 名患者接受ZOKINVY治疗,中位持续时间为 2.2 年,在推荐剂量 150 mg/m 2每天两次时约 1.9 年。人群为 2 至 17 岁,男性(33 [52%] 名患者)和女性(30 [48%] 名患者)的比例相似。与非经典 HGPS(2 [3%] 名患者)相比,大多数患者患有经典 HGPS(60 [95%] 名患者),1 名(2%)患者患有具有LMNA杂合突变的早老样椎板病。
表 3总结了临床试验中报告的不良反应。临床试验中最常见的不良反应(≥25%)是呕吐、腹泻、感染、恶心、食欲下降、疲劳、上呼吸道感染、腹痛、肌肉骨骼疼痛、电解质异常、体重减轻、头痛、骨髓抑制、增加天冬氨酸氨基转移酶、血碳酸氢盐降低、咳嗽、高血压和丙氨酸氨基转移酶升高。

胃肠道不良反应
如指出的表3中,胃肠道不良反应是最常报告的不良反应。在经历呕吐的 57 名患者中,30 名 (53%) 患者出现轻度呕吐(定义为无需干预),26 名 (46%) 患者出现中度呕吐(定义为门诊静脉补液;需要医疗干预)和 1 名(2 %) 患者有严重呕吐(定义为管饲、全胃肠外营养或住院)。在经历恶心的 35 名患者中,34 名 (97%) 患者有轻度恶心(定义为食欲不振但饮食习惯没有改变)和 1 名(3%)患者有中度恶心(定义为口服摄入量减少而体重没有显着下降,脱水或营养不良)。在研究 1 的前四个月治疗期间,19 名 (68%) 患者出现呕吐,10 名 (36%) 患者出现恶心。ZOKINVY需要止吐药或抗恶心药。共有 4 名患者停用ZOKINVY,主要是由于恶心或呕吐。在出现腹泻的 51 名患者中,大多数患者(约 92%)出现轻度或中度腹泻;38 (75%) 名患者报告轻度腹泻(定义为每天比基线增加少于 4 次)和 9 (18%) 名患者报告中度腹泻(定义为每天比基线增加 4 至 6 次;限制仪器日常生活活动)。四名 (8%) 患者报告严重腹泻(定义为每天大便次数超过基线 7 次或更多;需要住院治疗;与基线相比造口量严重增加;限制日常生活的自我护理活动)。在研究 1 的前四个月治疗期间,23 名 (82%) 患者出现腹泻;到治疗结束时,3 名 (11%) 患者出现腹泻。12 名 (43%) 患者接受了洛哌丁胺治疗。
丙氨酸氨基转移酶和天冬氨酸氨基转移酶升高
通常报告丙氨酸转氨酶升高(17 [27%] 名患者)。在丙氨酸转氨酶升高的 17 名患者中,14 名 (82%) 患者轻度升高(定义为大于正常上限 (ULN) 至 3.0 倍如果基线正常;如果基线异常则为 1.5 至 3.0 倍 ULN), 1 (6%) 名患者有中度增加(定义为大于 3.0 至 5.0 倍 ULN,如果基线正常或异常),2 名 (12%) 患者有严重增加(定义为大于 5.0 至 20.0 倍 ULN,如果基线正常或异常)正常或异常)。天冬氨酸转氨酶升高也经常被报道(22 [35%] 名患者)。在天冬氨酸转氨酶升高的 22 名患者中,21 名 (95%) 患者轻度升高(定义为大于 ULN 至 ULN 的 3.0 倍,如果基线正常;1.5 至 3。如果基线异常,则为 0 倍 ULN)和 1 (5%) 名患者出现严重增加(如果基线正常或异常,则定义为大于 5.0 至 20.0 倍 ULN)。一名丙氨酸和天冬氨酸转氨酶升高的患者也出现了高甘油三酯血症和高血糖,导致停药佐金维。高血压
用ZOKINVY治疗的患者有血压升高的记录。在基线时,22 名 (35%) 患者的收缩压或舒张压或两者均高于 95%。在试验过程中,18 名 (29%) 患者患有高血压,根据收缩压或舒张压测量值,在 3 次或更多情况下高于 95%。五名 (8%) 基线时血压正常的患者在治疗结束时收缩压或舒张压高于 95%。7 药物相互作用
7.1 其他药物对ZOKINVY 的影响
表 4显示了涉及影响ZOKINVY 的药物的临床显着药物相互作用。

7.2 ZOKINVY对其他药物的影响
表 5列出了涉及受ZOKINVY影响的药物的临床显着药物相互作用。

8 在特定人群中的使用
8.1 怀孕
风险摘要
基于动物研究发现,ZOKINVY当给予孕妇可引起胚胎-胎仔的危害。没有关于ZOKINVY在孕妇中使用的人类数据来评估主要出生缺陷、流产或不良母体或胎儿结局的药物相关风险。告知孕妇对胎儿的风险。在动物生殖研究中,在器官形成期间对妊娠大鼠口服 lonafarnib 产生胚胎-胎儿毒性,暴露量是人类在推荐剂量 150 mg/m 2每天两次的暴露量的 1.2 倍。在妊娠兔中,在器官形成期间口服 lonafarnib 产生骨骼畸形和变异,暴露量低于人类每天两次150 mg/m 2 的暴露量,母体毒性是人类每天两次150 mg/m 2暴露量的 26 倍(见数据)。
指示人群主要出生缺陷和流产的估计背景风险未知。所有怀孕都有出生缺陷、流产或其他不良后果的背景风险。在美国一般人群中,临床认可的妊娠中主要出生缺陷和流产的估计背景风险分别为 2% 至 4% 和 15% 至 20%。
数据
动物数据
在大鼠胚胎-胎儿发育研究中,在器官形成过程中口服 lonafarnib 导致植入后丢失(吸收)增加和胎儿体重和活胎儿数量减少,剂量为 30 mg/kg/天 (1.2乘以推荐剂量 150 mg/m 2每天两次的人体 AUC [血浆浓度-时间曲线下面积] )。在低于人体 AUC 150 mg/m 2每天两次时,未观察到对大鼠胚胎-胎儿发育的影响。在兔中,在器官形成期间口服 lonafarnib 导致骨骼畸形和全身暴露量的变化,在低于人 AUC 的推荐剂量 150 mg/m 2每天两次,以及母体毒性(体重减轻和流产)在 120 mg/ kg/天(每天两次150 mg/m 2 时人 AUC 的 26 倍)。
在大鼠的出生前和出生后发育研究中,在器官形成至哺乳期间,母体口服给药至多 20 mg/kg/天(AUC 低于人类 150 mg/m 2每天两次的AUC)未观察到对后代的影响。
8.2 泌乳
风险总结
没有关于ZOKINVY在人乳中的存在、对母乳喂养婴儿的影响或对产奶量的影响的数据。Lonafarnib 在大鼠乳汁中排泄(见数据)。当药物存在于动物乳中时,该药物很可能存在于人乳中。母乳喂养的发育和健康益处应与母亲对ZOKINVY的临床需求以及ZOKINVY或潜在母体状况对母乳喂养婴儿的任何潜在不利影响一起考虑。
数据
Lonafarnib 在哺乳期大鼠口服给药后从乳汁中排泄,12 小时时乳汁与血浆的平均浓度比为 1.5。8.3 具有生殖潜力的女性和男性
避孕
ZOKINVY当给予妊娠妇女时可导致胚胎-胎儿伤害[见在特殊人群中使用( 8.1 )]。忠告有生育潜力的女性在用ZOKINVY治疗期间使用适当有效的避孕措施。不孕
根据大鼠中的发现,ZOKINVY可能会降低具有生殖潜力的雌性和雄性的生育能力[见警告和注意事项 ( 5.5 ),非临床毒理学 ( 13.1 )]。8.4 儿科使用
ZOKINVY用于治疗 HGPS 和加工缺陷型早老性椎板病(具有早老蛋白样蛋白积累的杂合LMNA突变或纯合或复合杂合ZMPSTE24突变)的安全性和有效性已在 12 个月及以上的儿科患者中得到证实。在 2 岁及以上儿童患者中进行的充分和良好对照研究支持使用ZOKINVY用于这些适应症[见临床研究 ( 14 )]。
尚未确定ZOKINVY在 12 个月以下儿童患者中的安全性和有效性。
8.6 成人使用
ZOKINVY用于治疗 HGPS 和加工缺陷型早老性椎板病(具有具有早老蛋白样蛋白积累的杂合LMNA突变或纯合或复合杂合ZMPSTE24突变)的安全性和有效性已在成人中建立。在成人中使用ZOKINVY用于这些适应症是基于对 2 岁及以上儿童患者的充分和良好对照研究[见临床研究 ( 14 )]。
11 说明
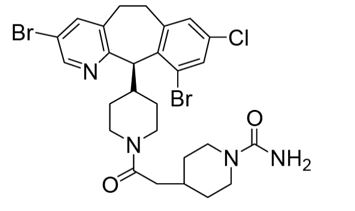
ZOKINVY (lonafarnib) 是一种法尼基转移酶抑制剂。lonafarnib 的化学名称是 4-[2-[4-[(11R)-3,10-dibromo-8-chloro-6,11-dihydro-5H-benzo[1,2] cyclohepta [2,4-b ]吡啶-11-基]哌啶-1-基]-2-氧乙基]哌啶-1-甲酰胺。其分子式为C 27 H 31 B r2 ClN 4 O 2,分子量为638.8 g/mol,其化学结构如下图所示。
用于口服给药的ZOKINVY(lonafarnib)胶囊含有 50 mg 或 75 mg lonafarnib 作为活性成分和以下非活性成分:交联羧甲基纤维素钠、硬脂酸镁、泊洛沙姆 188、聚维酮和二氧化硅。两种强度的胶囊壳均含有明胶、二氧化钛和氧化铁黄;75 毫克胶囊还含有红色氧化铁。压印油墨含有氨溶液、黑色氧化铁、丁醇、无水酒精、异丙醇、氢氧化钾、丙二醇、纯净水和虫胶。
12 临床药理学
12.1 作用机制
Lonafarnib 抑制法呢基转移酶,以防止法呢基化和随后的早老素和早老素样蛋白在内核膜中的积累。
12.2 药效学
尚未对ZOKINVY进行正式的药效学研究。
12.3 药代动力学
表 6总结了 lonafarnib 在 HGPS 患者中每日两次口服 lonafarnib 和食物后稳态时的药代动力学。

吸收
尚未确定口服给药后 lonafarnib 的绝对生物利用度。在空腹条件下,健康受试者每天两次口服 lonafarnib 75 mg 和 100 mg 后,lonafarnib 的几何平均 (CV%) 最大血浆峰浓度分别为 834 (32%) ng/mL 和 964 (32%) ng/mL , 分别。食物的影响
在健康受试者中单次口服 75 mg lonafarnib 后,与空腹条件相比,高脂肪餐(总热量 952 卡路里的约 43% 的脂肪)的 C max降低了 55%,AUC 降低了 29%。与禁食条件相比,低脂餐(总热量 421 卡路里的约 12% 的脂肪)的C max降低了 25%,AUC 降低了 21%。分布
在 0.5 至 40.0 μg/mL 的浓度范围内,lonafarnib 的体外血浆蛋白结合率大于或等于 99%。在健康受试者中每日两次口服 lonafarnib 100 mg 和 75 mg 后,稳态表观分布容积分别为 87.8 L 和 97.4 L。消除
在健康受试者中口服 lonafarnib 100 mg 每天两次后平均半衰期约为 4 至 6 小时。代谢
Lonafarnib 在体外主要由 CYP3A 代谢,并在较小程度上由 CYP1A2、CYP2A6、CYP2C8、CYP2C9、CYP2C19 和 CYP2E1 代谢。排泄健康受试者在禁食条件下
口服 104 mg [ 14 C]-lonafarnib 后,大约 62% 的总放射性标记剂量在粪便中回收,<1% 的总放射性标记剂量在 240 小时内从尿液中回收给药后。两种最主要的代谢物是 HM17 和 HM21(一种活性代谢物),分别占血浆放射性的 15% 和 14%。特定人群
肾受损或肝受损患者
ZOKINVY尚未在肾受损患者或肝受损患者中进行研究。男性和女性患者
在健康受试者中单次口服剂量 100 mg lonafarnib 后,与男性受试者相比,女性受试者的血浆 lonafarnib AUC 和 C max分别高 44% 和 26%。在健康受试者中观察到的不同性别的暴露差异被认为没有临床意义。老年患者
在健康受试者中单次口服 100 mg lonafarnib 后,与 18 至 45 岁受试者相比,≥65 岁受试者的血浆 lonafarnib 暴露 AUC 和Cmax分别高 59% 和 27%。在老年受试者中观察到的较高暴露被认为与临床无关。药物相互作用研究
体外研究
Lonafarnib 是一种 CYP3A 底物和一种有效的 CYP3A 时间依赖性和基于机制的抑制剂。Lonafarnib 是 CYP2C8 和 CYP2C19 的抑制剂。Lonafarnib 不被认为是 CYP1A2、CYP2B6、CYP2C8、CYP2C9 或 CYP2D6 的抑制剂。Lonafarnib 不太可能是 CYP1A2、CYP2B6 和 CYP3A 的诱导剂。Lonafarnib 不是转运蛋白 OATP1B1、OATP1B3 或 BCRP 的底物,但可能是 P-gp 的边缘底物。Lonafarnib 是 P-gp、OATP1B1、OATP1B3 和 BCRP 的抑制剂。
临床研究:其他药物对 Lonafarnib 的影响
CYP3A 抑制剂
Lonafarnib 是CYP3A的敏感底物。在 200 mg 酮康唑(一种强效 CYP3A 抑制剂)每天 1 次共 5 天后,单次口服 50 mg lonafarnib与 lonafarnib 相比,lonafarnib的 C max和 AUC 分别增加了 270% 和 425%在健康受试者中单独使用[见剂量和给药方法 ( 2.3 ),禁忌症 ( 4 ),药物相互作用 ( 7.1 )]。CYP2C9 抑制剂
与CYP2C9 抑制剂共同给药可能会增加 lonafarnib AUC 和Cmax。尚未进行ZOKINVY与 CYP2C9 抑制剂的药物-药物相互作用研究[见药物相互作用( 7.1 )]。CYP3A 诱导剂
单次口服 50 mg lonafarnib(联合单次口服 100 mg 利托那韦)后,每天服用 600 mg 利福平,持续 8 天,lonafarnib的 C max降低 92%,AUC 降低与在健康受试者中未联合使用利福平相比,降低了 98% [见禁忌症 ( 4 ),药物相互作用 ( 7.1 )]。临床研究:Lonafarnib 对其他药物的影响
CYP3A 底物
Lonafarnib 是CYP3A的强抑制剂。在健康受试者中,单次口服 3 mg 咪达唑仑与多次口服 100 mg lonafarnib 每日两次共给药 5 天,咪达唑仑的 C max和 AUC 分别增加了 180% 和 639% [见剂量和给药方法] ( 2.4 )、禁忌症 ( 4 )、药物相互作用 ( 7.2 )]。洛哌丁胺
在健康受试者中,单次口服 2 mg 洛哌丁胺(主要由 CYP2C8 和 CYP3A 代谢以及 P-gp 的底物)与多次口服 lonafarnib 100 mg 每天两次共 5 天共同给药,Cmax和 AUC 为洛哌丁胺分别增加了 214% 和 299% [见药物相互作用 ( 7.2 )]。CYP2C19 底物
Lonafarnib 是一种中度 CYP2C19 抑制剂。在健康受试者中,单次口服 40 mg 奥美拉唑与多次口服 lonafarnib 75 mg 每天两次共给药 5 天,奥美拉唑的 C max和 AUC 分别增加了 28% 和 60% [见药物相互作用(7.2 )]。P-gp 和 OATP1B 底物
在健康受试者中,将单次口服剂量的 180 mg 非索非那定(一种 P-gp 和 OATP1B 底物)与多次口服剂量的 100 mg lonafarnib 每天两次共给药 5 天,非索非那定的Cmax和 AUC分别增加了 21% 和 24% [见药物相互作用 ( 7.2 )]。13 非临床毒理学
13.1 致癌作用、诱变作用、生育力受损
致癌作用
尚未对 lonafarnib 进行致癌性研究。诱变
Lonafarnib 在细菌致突变性 (Ames) 试验、哺乳动物细胞体外染色体畸变试验或小鼠体内微核试验中没有遗传毒性。生育力
受损 Lonafarnib 在雄性大鼠中在 90 mg/kg/day 或更高时产生生育力受损(推荐剂量为 150 mg/m 2每天两次的人类 AUC 的 1.5 倍),在 180 mg 时几乎完全丧失生育力/kg/天(人类 AUC 的 3 倍)。用 180 mg/kg/天治疗的雄性大鼠表现出睾丸小、睾丸松弛和附睾变色(分别为雄性的 84%、56% 和 24%)。男性在全身暴露量低于 150 mg/m 2每天两次的人 AUC 时对生育力没有影响。用 30 mg/kg/day lonafarnib 或更高剂量(推荐人用剂量 150 mg/m 2每天两次时人 AUC 的 1.2 倍)治疗的雌性大鼠显示生育力下降,表现为黄体数量减少和植入部位,以及植入前和植入后损失的增加。女性在全身暴露低于 150 mg/m 2每天两次的人 AUC 时对生育力没有影响[见警告和注意事项 ( 5.5 )]。
13.2 动物毒理学和/或药理学
在一项为期 6 个月的 lonafarnib 大鼠口服毒性研究中发生肾毒性,在全身暴露时观察到肾脏病变(间质坏死和内髓质矿化)和相关的临床化学变化(例如高磷血症、高钾血症)和尿液分析参数等于推荐剂量 150 mg/m 2每天两次的人体 AUC 。在低于人 AUC 150 mg/m 2每天两次全身暴露时未观察到肾毒性证据[见警告和注意事项( 5.3 )]。
在猴子的 1 年口服毒性研究中,雄性生殖道毒性为 10 mg/kg/天或更高(AUC 低于推荐剂量 150 mg/m 2每天两次的人 AUC )。男性生殖道的病变包括附睾无精子和生精小管、精囊和前列腺的萎缩。在大鼠中也观察到睾丸毒性,其中雄性生育能力严重受损[见警告和注意事项 ( 5.5 ),非临床毒理学 ( 13.1 )]。
在猴子的 1 年口服毒性研究中发生了眼(视网膜)毒性,剂量为 40 mg/kg/天(推荐剂量为 150 mg/m 2每天两次时人体 AUC 的 3.7 倍)。视网膜损伤涉及视杆细胞层和视锥细胞层和外核层中感光细胞的单细胞坏死。在 20 mg/kg/天时未观察到视网膜毒性(150 mg/m 2 时人 AUC 的 2.1 倍)每天两次)。然而,在通过视网膜电图评估 lonafarnib 对猴子视觉功能影响的后续研究中,口服 15 mg/kg/天共 13 周或 60 mg/kg/天共 6 周对视杆依赖性, 弱光视力。在整个治疗期间的几个时间点观察到效果。在研究终止时未观察到视网膜组织学变化[见警告和注意事项( 5.4 )]。
14 临床研究
ZOKINVY的疗效基于观察性队列生存研究的结果,该研究回顾性比较了 HGPS 患者与自然病史队列的两项 2 期研究的生存数据。
研究 1 (NCT00425607) 是一项 2 期开放标签、单臂试验,评估了ZOKINVY在 28 名患者中的疗效(26 名具有经典 HGPS,1 名具有非经典 HGPS,1 名患有具有早老素的LMNA杂合突变的早老性椎板病- 像蛋白质积累)。患者接受ZOKINVY治疗 24 至 30 个月。患者开始用ZOKINVY 115 mg/m 2每天两次治疗。治疗4个月后,耐受治疗的患者剂量增加至150 mg/m 2每天两次。在接受治疗的 28 名患者中,27 名 HGPS 患者(16 名女性,11 名男性)被纳入生存评估。27 名患者开始治疗时的中位年龄为 7.5 岁(范围:3 至 16 岁)。体重范围为 6.6 至 17.6 kg,BSA 范围为 0.38 至 0.75 m 2(ZOKINVY不适用于 BSA 小于 0.39 m 2 的患者,因为该人群没有合适的剂量强度)。
研究 1 完成后,26 名患者参加了第二个 2 期开放标签单臂试验(研究 2,NCT00916747),该试验由两个研究阶段组成。在研究 2 的第一阶段,患者接受ZOKINVY和额外治疗约 5 年。在研究 2 的第二阶段,患者接受ZOKINVY 150 mg/m 2每天两次,持续长达 3 年。
有 35 名 HGPS 初治患者被纳入研究 2 的第二阶段。 在 35 名接受治疗的患者(22 名男性,13 名女性)中,34 名 (97.1%) 患者患有经典 HGPS,1 名 (2.9%) 患者患有非经典全球定位系统。中位年龄为 6 岁(范围:2 至 17 岁)。体重范围为 6.7 至 22 kg,BSA 范围为 0.42 至 0.90 m 2。
在整个研究 1 和研究 2 中,ZOKINVY通过胶囊口服给药,或者将胶囊内容物与 Ora Blend SF 或 Ora-Plus 混合并作为悬浮液口服给药。
回顾性生存分析基于来自 62 名接受治疗的患者(研究 1 中的 27 名患者和研究 2 中的 35 名初治患者)的死亡率数据以及来自单独自然史队列中匹配的未治疗患者的数据。与治疗的患者HGPS的寿命ZOKINVY通过比未经治疗的患者最后的随访时间(11岁)通过前三年的随访和2.5年增加了平均3个月。生存分析总结在表7和图1中提供。表 7:HGPS 患者的生存分析总结

图 1:HGPS 患者在最后一次随访时截尾的随访时间的 Kaplan-Meier 生存曲线
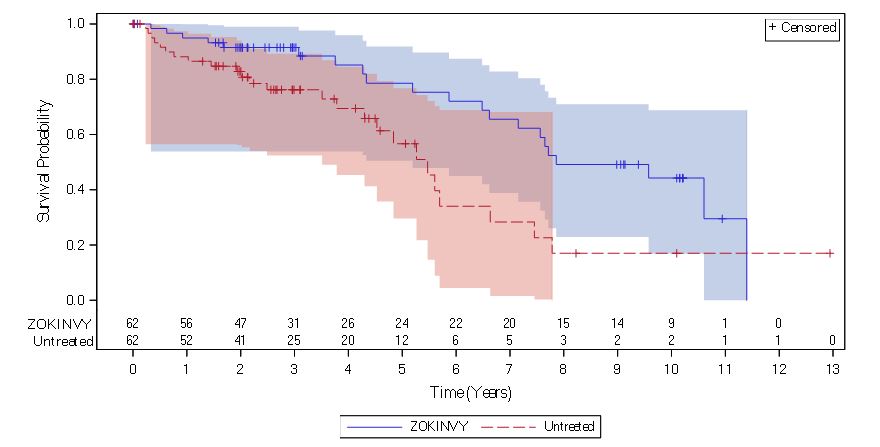
注:ZOKINVY治疗患者的 Kaplan-Meier (KM) 生存曲线用实线表示;未治疗患者的曲线用虚线表示。蓝色和红色阴影区域分别代表治疗和未治疗 KM 生存曲线的 95% 置信带。
16 如何供应/储存和处理
ZOKINVY提供为:
- 50 毫克胶囊:4 号硬胶囊,不透明黄色,“LNF”和“50”印有黑色。
每瓶 30 粒胶囊 (NDC 73079-050-30) - 75 毫克胶囊:3 号硬胶囊,不透明浅橙色,黑色印有“LNF”和“75”。
每瓶 30 粒胶囊 (NDC 73079-075-30)
储存在 20°C-25°C (68°F-77°F),允许在 15°C-30°C (59°F-86°F) 下游览 [参见 USP 控制室温]。
17 患者咨询信息
. 建议患者阅读 FDA 批准的患者标签(患者信息和使用说明)。
给药[见剂量和给药方法( 2.1 )]
- 忠告患者和护理人员ZOKINVY应每天服用两次,随早晚饭一起服用。
- 告知患者和护理人员,如果错过剂量,应尽快在下一次计划剂量前 8 小时给予下一剂。如果距离下一次预定剂量还有不到 8 小时,患者应跳过错过的剂量并在下一次预定剂量继续服用ZOKINVY
制备和给药[见剂量和给药方法 ( 2.5 ),药物相互作用 ( 7 )]
- 建议患者用水吞服整个胶囊。不应咀嚼胶囊。
- 对于无法吞咽胶囊的患者,建议患者和护理人员将ZOKINVY的内容物与 Ora Blend SF 或 Ora-Plus 混合。对于无法获得或耐受 Ora Blend SF 或 Ora-Plus 的患者,ZOKINVY的内容物可以与橙汁或苹果酱混合。忠告患者不要将ZOKINVY的内容物与含有葡萄柚或塞维利亚橙子的果汁混合。建议患者和护理人员每次剂量必须新鲜制备混合物,并在混合后约 10 分钟内服用。
- 建议患者和护理人员阅读并仔细遵循使用 Ora Blend SF、Ora-Plus、橙汁或苹果酱中的胶囊内容物的说明[见使用说明]。建议患者和护理人员在有任何问题时致电他们的医疗保健提供者或药剂师。
药物相互作用[见剂量和给药方法 ( 2.3 , 2.4 ),禁忌症 ( 4 ),警告和注意事项 ( 5.1 ),药物相互作用 ( 7 )]
告知患者和护理人员ZOKINVY可能与许多药物相互作用。建议患者及其护理人员报告患者使用所有处方药和非处方药的情况,包括营养补充剂和维生素。胃肠道不良反应[见剂量和给药方法( 2.2 ),不良反应( 6.1 )]
告知患者和护理人员胃肠道不良反应在ZOKINVY 中很常见。这些包括但不限于呕吐、腹泻和恶心。如果这些不良反应持续存在,建议患者和护理人员联系他们的医疗保健提供者。高血压[见不良反应 ( 6.1 )]
告知患者和护理人员服用ZOKINVY 时血压可能升高。高血压的症状可能包括头痛、气短、流鼻血、脸红、头晕或胸痛。如果发生这些不良反应,建议患者和护理人员联系他们的医疗保健提供者。肾毒性[见警告和注意事项 ( 5.3 ),非临床毒理学 ( 13.2 )]
告知患者和护理人员肾损伤的风险。视网膜毒性[见警告和注意事项 ( 5.4 ),非临床毒理学 ( 13.2 )]
告知患者和护理人员发生夜视困难的风险。建议患者和护理人员在视力发生变化时联系他们的医疗保健提供者。生育能力受损[见警告和注意事项 ( 5.5 ),非临床毒理学 ( 13.1 )]
告知女性和男性生殖潜能ZOKINVY可能影响青春期发育和损害生育能力。胚胎-胎儿毒性[见警告和注意事项 ( 5.6 ),在特殊人群中使用 ( 8.1 , 8.3 )]
告知孕妇和女性患者对胎儿的潜在风险的生殖潜能。忠告有生育潜力的女性在用ZOKINVY治疗期间使用有效的避孕措施。制造地:
Eiger BioPharmaceuticals, Inc., 2155 Park Boulevard Palo Alto, CA 94306
ZOKINVY是 Eiger BioPharmaceuticals, Inc. 的商标。

主要显示面板 - NDC:73079-050-30 - 50 毫克 30 支瓶标签

主要显示面板 - NDC:73079-050-30 - 50 毫克 30 支瓶纸箱标签

主要显示面板 - NDC:73079-075-30 - 75 毫克 30 支瓶标签

主要显示面板 - NDC:73079-075-30 - 75 毫克 30 支瓶纸箱标签

【备注】以上内容仅供参考,不作为用药依据,详情请参照药品附带说明书。
-
本说明书来源于:美国FDA
温馨提醒:
①建议您用 谷歌浏览器 在电脑上或手机 打开以上链接,就可以自动翻译成简体中文,而且翻译的还比较准确。
②本说明书仅供参考,最新的说明书详见药品附带的说明书
1 INDICATIONS AND USAGE
ZOKINVY is indicated in patients 12 months of age and older with a body surface area (BSA) of 0.39 m2 and above:
- To reduce the risk of mortality in Hutchinson-Gilford Progeria Syndrome (HGPS)
- For the treatment of processing-deficient Progeroid Laminopathies with either:
- Heterozygous LMNA mutation with progerin-like protein accumulation
- Homozygous or compound heterozygous ZMPSTE24 mutations
Limitations of Use
ZOKINVY is not indicated for other Progeroid Syndromes or processing-proficient Progeroid Laminopathies. Based upon its mechanism of action, ZOKINVY would not be expected to be effective in these populations.
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
- The starting dosage of ZOKINVY for patients with a BSA of 0.39 m2 and above is 115 mg/m2 twice daily with morning and evening meals (see Table 1) to reduce the risk of gastrointestinal adverse reactions [see Adverse Reactions (6.1)]. An appropriate dosage strength of ZOKINVY is not available for patients with a BSA of less than 0.39 m2 [see Indications and Usage (1)].
- After 4 months of treatment, increase the dosage to 150 mg/m2 twice daily with morning and evening meals (see Table 2).
- Round all total daily dosages to the nearest 25 mg increment (see Table 1 and Table 2).
- If a dose is missed, take the dose as soon as possible with food, up to 8 hours prior to the next scheduled dose. If less than 8 hours remains before the next scheduled dose, skip the missed dose, and resume taking ZOKINVY at the next scheduled dose.
Table 1 provides the BSA-based dosage recommendations for the starting dosage of 115 mg/m2 twice daily.

Table 2 provides the BSA-based dosage recommendations for the dosage of 150 mg/m2 twice daily.

2.2 Dosage Modifications for Gastrointestinal Adverse Reactions
For patients who have increased their dose of ZOKINVY to 150 mg/m2 twice daily and are experiencing repeated episodes of vomiting and/or diarrhea resulting in dehydration or weight loss, ZOKINVY can be dose reduced to the starting dose of 115 mg/m2 twice daily (see Table 1). Ensure ZOKINVY is taken with the morning and evening meals and an adequate amount of water.
2.3 Dosage Modifications for Drug Interactions
CYP3A Inhibitors
If concomitant use of ZOKINVY with a weak CYP3A inhibitor is unavoidable [see Warnings and Precautions (5.1), Drug Interactions (7.1)]:- Reduce to or continue ZOKINVY at the starting dosage of 115 mg/m2 twice daily (see Table 1).
- Resume the previous ZOKINVY dosage 14 days after discontinuing the concomitant use of the weak CYP3A inhibitor.
2.4 Temporary Discontinuation for Midazolam Use
Temporarily discontinue ZOKINVY for 10 to 14 days before and 2 days after administration of midazolam [see Contraindications (4), Drug Interactions (7.2)].
2.5 Preparation and Administration Instructions
Administer ZOKINVY orally with the morning and evening meals.
Patients Able to Swallow Capsules
- Administer ZOKINVY capsules whole with a sufficient amount of water. Do not chew the capsules.
Patients Unable to Swallow Capsules
- The entire contents of ZOKINVY capsules can be mixed with Ora Blend SF® or Ora-Plus® or, for patients unable to access or tolerate Ora Blend SF or Ora-Plus, the contents of ZOKINVY capsules can be mixed with orange juice or applesauce (see preparation instructions below).
- Do not mix with juice containing grapefruit or Seville oranges [see Contraindications (4), Drug Interactions (7.1)].
- The mixture must be prepared fresh for each dose and be taken within approximately 10 minutes of mixing.
Preparation of Dose in Ora Blend SF, Ora-Plus, or Orange Juice
- For each capsule, empty contents of the capsule into a container containing 5 mL to 10 mL of the liquid.
- Mix thoroughly with a spoon.
- Consume entire serving.
Preparation of Dose in Applesauce
- For each capsule, empty contents of the capsule into a container containing 1 teaspoonful to 2 teaspoonfuls of applesauce.
- Mix thoroughly with a spoon.
- Consume entire serving.
3 DOSAGE FORMS AND STRENGTHS
Capsules:
- 50 mg, opaque yellow with “LNF” and “50” printed in black
- 75 mg, opaque light orange with “LNF” and “75” printed in black
4 CONTRAINDICATIONS
ZOKINVY is contraindicated in patients taking:
- Strong or moderate CYP3A inhibitors or inducers [see Drug Interactions (7.1)]
- Midazolam [see Drug Interactions (7.2)]
- Lovastatin, simvastatin, or atorvastatin [see Drug Interactions (7.2)]
5 WARNINGS AND PRECAUTIONS
5.1 Risk of Reduced Efficacy or Adverse Reactions Due to Drug Interactions
Coadministration of ZOKINVY with other drugs may result in clinically significant drug interactions [see Dosage and Administration (2.3, 2.4), Contraindications (4), Drug Interactions (7.1, 7.2)]. These drug interactions can lead to:
- Reduced efficacy of ZOKINVY
- Increased risk of adverse reactions from ZOKINVY or co-administered drugs
See Table 4 and Table 5 for steps to prevent or manage these clinically significant drug interactions, including dosage recommendations [see Drug Interactions (7.1, 7.2)]. Consider the potential for drug interactions prior to and during ZOKINVY therapy; review concomitant medications during ZOKINVY therapy; and monitor for the adverse reactions.
5.2 Laboratory Abnormalities
Some patients treated with ZOKINVY developed laboratory abnormalities [see Adverse Reactions (6.1)]. These included:
- Electrolyte abnormalities (43%), such as hyperkalemia, hypokalemia, hyponatremia, or hypercalcemia
- Myelosuppression (35%), such as reductions in absolute neutrophil count, white blood cell counts, lymphocytes, hemoglobin, or hematocrit
- Increased liver enzymes, such as aspartate aminotransferase (35%), or alanine aminotransferase (27%)
These laboratory abnormalities often improved while continuing ZOKINVY, but it is not possible to exclude ZOKINVY as a cause of the abnormalities. Periodically monitor electrolytes, complete blood counts, and liver enzymes, and manage abnormalities accordingly.
5.3 Nephrotoxicity
Lonafarnib caused nephrotoxicity in rats at plasma drug exposures approximately equal to that achieved with the human dose [see Nonclinical Toxicology (13.2)]. Monitor renal function at regular intervals during ZOKINVY therapy.
5.4 Retinal Toxicity
Lonafarnib caused rod-dependent, low-light vision decline in monkeys at plasma drug exposures similar to that achieved with the human dose [see Nonclinical Toxicology (13.2)]. Perform ophthalmological evaluation at regular intervals and at the onset of any new visual changes during ZOKINVY therapy.
5.5 Impaired Fertility
Lonafarnib caused impaired fertility in female rats at 1.2 times the human dose based on plasma drug exposure [see Nonclinical Toxicology (13.1)].
Lonafarnib caused impaired fertility and testicular toxicity in male rats at 1.5 times the human dose based on plasma drug exposure [see Nonclinical Toxicology (13.1)], and toxicity in the male reproductive tract in monkeys at doses lower than the human dose based on plasma drug exposure [see Nonclinical Toxicology (13.2)].
Advise females and males of reproductive potential of the animal fertility findings, and that the impact on pubertal development and the potential for impaired fertility with ZOKINVY therapy in humans have not been adequately evaluated [see Use in Specific Populations (8.3)].
5.6 Embryo-Fetal Toxicity
Based on findings from animal reproduction studies, ZOKINVY can cause embryo-fetal harm when administered to pregnant women. In animal reproduction studies, oral administration of lonafarnib in pregnant rats during organogenesis produced embryo-fetal toxicity at plasma drug exposures that were approximately equal to the recommended human dose. In pregnant rabbits, oral administration of lonafarnib during organogenesis produced skeletal malformations and variations at exposures lower than the human exposure. Advise pregnant women of the risk to a fetus. Advise females of reproductive potential to avoid becoming pregnant and to use appropriate effective contraception during treatment with ZOKINVY [see Use in Specific Populations (8.1, 8.3)].
6 ADVERSE REACTIONS
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
A total of 84 subjects were treated with at least one dose of ZOKINVY with or without additional therapy, of which 8 were treated at a dosage of at least 115 mg/m2 twice daily for greater than or equal to 10 years.
The safety profile of ZOKINVY is based on 128 patient-years of treatment exposure (62 patients with HGPS and 1 patient with processing-deficient Progeroid Laminopathy with LMNA heterozygous mutation) and pooled results from two Phase 2 open-label, single-arm trials (n=63: 28 patients from Study 1 and 35 treatment naïve patients from Study 2). In Study 1, ZOKINVY treatment was initiated at 115 mg/m2 twice daily and increased to 150 mg/m2 twice daily after approximately 4 months for a total treatment duration of 24 to 30 months. Treatment naïve patients in Study 2 received ZOKINVY 150 mg/m2 twice daily for up to 36 months. In both studies, ZOKINVY was administered orally via capsules or the capsule contents were mixed with Ora Blend SF or Ora-Plus and administered orally as a suspension.
In these two studies, a total of 63 patients received ZOKINVY for a median duration of 2.2 years, with approximately 1.9 years at the recommended dose of 150 mg/m2 twice daily. The population was 2 to 17 years old, with a similar proportion of males (33 [52%] patients) and females (30 [48%] patients). Most patients had classic HGPS (60 [95%] patients) compared to non-classic HGPS (2 [3%] patients) and 1 (2%) patient had Progeroid Laminopathy with LMNA heterozygous mutation.
Table 3 summarizes adverse reactions reported in the clinical trials. The most common adverse reactions (≥25%) in the clinical trials were vomiting, diarrhea, infection, nausea, decreased appetite, fatigue, upper respiratory tract infection, abdominal pain, musculoskeletal pain, electrolyte abnormalities, decreased weight, headache, myelosuppression, increased aspartate aminotransferase, decreased blood bicarbonate, cough, hypertension, and increased alanine aminotransferase.

Gastrointestinal Adverse Reactions
As noted in Table 3, gastrointestinal adverse reactions were the most frequently reported adverse reactions. Of the 57 patients who experienced vomiting, 30 (53%) patients had mild vomiting (defined as no intervention required), 26 (46%) patients had moderate vomiting (defined as outpatient intravenous hydration; medical intervention required), and 1 (2%) patient had severe vomiting (defined as tube feeding, total parental nutrition, or hospitalization indicated). Of the 35 patients who experienced nausea, 34 (97%) patients had mild nausea (defined as loss of appetite without alteration in eating habits) and 1 (3%) patient had moderate nausea (defined as oral intake decreased without significant weight loss, dehydration, or malnutrition). During the first four months of treatment in Study 1, 19 (68%) patients had vomiting and 10 (36%) patients had nausea. By the end of therapy, 4 (14%) patients who were still on ZOKINVY required antiemetics or anti-nauseants. A total of 4 patients discontinued ZOKINVY, mostly due to nausea or vomiting.Of the 51 patients who experienced diarrhea, the majority of patients (approximately 92%) experienced mild or moderate diarrhea; 38 (75%) patients reported mild diarrhea (defined as an increase of less than 4 stools per day over baseline) and 9 (18%) patients reported moderate diarrhea (defined as increase of 4 to 6 stools per day over baseline; limiting instrumental activities of daily living). Four (8%) patients reported severe diarrhea (defined as increase of seven or more stools per day over baseline; hospitalization indicated; severe increase in ostomy output compared to baseline; limiting self-care activities of daily living). During the first four months of treatment in Study 1, 23 (82%) patients had diarrhea; by the end of therapy, 3 (11%) patients had diarrhea. Twelve (43%) patients were treated with loperamide.
Alanine Aminotransferase and Aspartate Aminotransferase Elevations
Increased alanine aminotransferase was commonly reported (17 [27%] patients). Of the 17 patients with increased alanine aminotransferase, 14 (82%) patients had mild increases (defined as greater than upper limit of normal (ULN) to 3.0 times ULN if baseline was normal; 1.5 to 3.0 times ULN if baseline was abnormal), 1 (6%) patient had moderate increases (defined as greater than 3.0 to 5.0 times ULN if baseline was normal or abnormal), and 2 (12%) patients had severe increases (defined as greater than 5.0 to 20.0 times ULN if baseline was normal or abnormal). Increased aspartate aminotransferase was also commonly reported (22 [35%] patients). Of the 22 patients with increased aspartate aminotransferase, 21 (95%) patients had mild increases (defined as greater than ULN to 3.0 times ULN if baseline was normal; 1.5 to 3.0 times ULN if baseline was abnormal) and 1 (5%) patient had a severe increase (defined as greater than 5.0 to 20.0 times ULN if baseline was normal or abnormal). One patient with alanine and aspartate aminotransferase elevations also experienced hypertriglyceridemia and hyperglycemia resulting in discontinuation of ZOKINVY.Hypertension
Increases in blood pressure have been documented in patients treated with ZOKINVY. At baseline 22 (35%) patients had either a systolic blood pressure or a diastolic blood pressure or both above the 95th percentile. Over the course of the trials, 18 (29%) patients had hypertension based on systolic blood pressure or diastolic blood pressure measurements above the 95th percentile on 3 or more occasions. Five (8%) patients who were normotensive at baseline had either systolic blood pressure or diastolic blood pressure above the 95th percentile at the end of treatment.7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on ZOKINVY
Table 4 presents clinically significant drug interactions involving drugs that affect ZOKINVY.

7.2 ZOKINVY’s Effect on Other Drugs
Table 5 presents clinically significant drug interactions involving drugs affected by ZOKINVY.

8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal studies, ZOKINVY can cause embryofetal harm when administered to a pregnant woman. There are no human data on ZOKINVY use in pregnant women to evaluate for a drug-associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes. Advise pregnant women of the risk to a fetus.In animal reproduction studies, oral administration of lonafarnib to pregnant rats during organogenesis produced embryo-fetal toxicity at exposures that were 1.2-times the human exposure at the recommended dose of 150 mg/m2 twice daily. In pregnant rabbits, oral administration of lonafarnib during organogenesis produced skeletal malformations and variations at exposures lower than the human exposure at 150 mg/m2 twice daily, and maternal toxicity at 26 times the human exposure at 150 mg/m2 twice daily (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Animal Data
In an embryo-fetal development study in rats, oral administration of lonafarnib during organogenesis produced an increase in post-implantation loss (resorptions) and decreases in fetal body weight and number of live fetuses at 30 mg/kg/day (1.2 times the AUC [area under the plasma concentration-time curve] in humans at the recommended dose of 150 mg/m2 twice daily). No effects on embryo-fetal development in rats were observed at systemic exposures lower than the human AUC at 150 mg/m2 twice daily.In rabbits, oral administration of lonafarnib during organogenesis resulted in skeletal malformations and variations at systemic exposures lower than the human AUC at the recommended dose of 150 mg/m2 twice daily, and maternal toxicity (body weight loss and abortion) at 120 mg/kg/day (26 times the human AUC at 150 mg/m2 twice daily).
No effects in offspring were observed in a pre- and postnatal development study in rats with maternal administration of up to 20 mg/kg/day orally (AUC lower than the human AUC at 150 mg/m2 twice daily) during organogenesis through lactation.
8.2 Lactation
Risk Summary
There are no data on the presence of ZOKINVY in human milk, the effects on the breastfed infant, or the effects on milk production. Lonafarnib is excreted in rat milk (see Data). When a drug is present in animal milk, it is likely that the drug will be present in human milk.The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for ZOKINVY and any potential adverse effects of the breastfed infant from ZOKINVY or from the underlying maternal condition.
Data
Lonafarnib is excreted in milk following oral administration in lactating rats, with a mean milk to plasma concentration ratio of 1.5 at 12 hours.8.3 Females and Males of Reproductive Potential
Contraception
ZOKINVY can cause embryo-fetal harm when administered to pregnant women [see Use in Specific Populations (8.1)]. Advise females of reproductive potential to use appropriate effective contraception during treatment with ZOKINVY.Infertility
Based on findings in rats, ZOKINVY may reduce fertility in females and males of reproductive potential [see Warnings and Precautions (5.5), Nonclinical Toxicology (13.1)].8.4 Pediatric Use
The safety and effectiveness of ZOKINVY for the treatment of HGPS and processing-deficient Progeroid Laminopathies (with either heterozygous LMNA mutation with progerin-like protein accumulation or homozygous or compound heterozygous ZMPSTE24 mutations) have been established in pediatric patients 12 months of age and older. Use of ZOKINVY for these indications is supported by adequate and well controlled studies in pediatric patients 2 years of age and older [see Clinical Studies (14)].
The safety and effectiveness of ZOKINVY in pediatric patients less than 12 months of age have not been established.
8.6 Adult Use
The safety and effectiveness of ZOKINVY for the treatment of HGPS and processing-deficient Progeroid Laminopathies (with either heterozygous LMNA mutation with progerin-like protein accumulation or homozygous or compound heterozygous ZMPSTE24 mutations) have been established in adults. Use of ZOKINVY in adults for these indications is based on adequate and well controlled studies in pediatric patients 2 years of age and older [see Clinical Studies (14)].
11 DESCRIPTION
ZOKINVY (lonafarnib) is a farnesyltransferase inhibitor. The chemical name for lonafarnib is 4-[2-[4-[(11R)-3,10-dibromo-8-chloro-6,11-dihydro-5H- benzo[1,2] cyclohepta [2,4-b]pyridin-11-yl]piperidin-1-yl]-2- oxoethyl]piperidine-1-carboxamide. Its molecular formula is C27H31Br2ClN4O2, molecular mass is 638.8 g/mol, and its chemical structure is depicted below.
ZOKINVY (lonafarnib) capsules for oral administration contain 50 mg or 75 mg of lonafarnib as the active ingredient and the following inactive ingredients: croscarmellose sodium, magnesium stearate, poloxamer 188, povidone, and silicon dioxide. The capsule shells of both strengths contain gelatin, titanium dioxide, and yellow iron oxide; the 75 mg capsule also contains red iron oxide. The imprinting ink contains ammonia solution, black iron oxide, butyl alcohol, dehydrated alcohol, isopropyl alcohol, potassium hydroxide, propylene glycol, purified water, and shellac.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Lonafarnib inhibits farnesyltransferase to prevent farnesylation and subsequent accumulation of progerin and progerin-like proteins in the inner nuclear membrane.
12.2 Pharmacodynamics
No formal pharmacodynamic studies have been conducted with ZOKINVY.
12.3 Pharmacokinetics
The pharmacokinetics of lonafarnib at steady state in patients with HGPS following oral administration of lonafarnib twice daily with food are summarized in Table 6.

Absorption
The absolute bioavailability of lonafarnib following oral administration has not been determined. Following oral administration of lonafarnib 75 mg and 100 mg twice daily in healthy subjects under fasted conditions, the geometric mean (CV%) maximum peak plasma concentrations of lonafarnib were 834 (32%) ng/mL and 964 (32%) ng/mL, respectively.Effect of Food
Following a single oral dose of 75 mg lonafarnib in healthy subjects, the Cmax decreased 55% and AUC decreased 29% with a high-fat meal (approximately 43% fat of the total 952 calories) compared to fasted conditions. Cmax decreased 25% and AUC decreased 21% with a low-fat meal (approximately 12% fat of the total 421 calories) compared to fasted conditions.Distribution
In vitro plasma protein binding of lonafarnib was greater than or equal to 99% over the concentration range between 0.5 to 40.0 μg/mL. The apparent volumes of distribution were 87.8 L and 97.4 L, respectively, at steady state following oral administration of lonafarnib 100 mg and 75 mg twice daily in healthy subjects.Elimination
The mean half-life was approximately 4 to 6 hours following oral administration of lonafarnib 100 mg twice daily in healthy subjects.Metabolism
Lonafarnib is primarily metabolized by CYP3A and to a lesser extent by CYP1A2, CYP2A6, CYP2C8, CYP2C9, CYP2C19, and CYP2E1 in vitro.Excretion
Following an oral administration of 104 mg [14C]-lonafarnib under fasted conditions in healthy subjects, approximately 62% of the total radiolabeled dose was recovered in feces and <1% of the total radiolabeled dose was recovered in urine up to 240 hours post-dose. The two most predominant metabolites were HM17 and HM21 (an active metabolite) accounting for 15% and 14% of plasma radioactivity, respectively.Specific Populations
Patients with Renal Impairment or Hepatic Impairment
ZOKINVY has not been studied in patients with renal impairment or in patients with hepatic impairment.Male and Female Patients
Following a single oral dose of 100 mg lonafarnib in healthy subjects, the plasma lonafarnib AUC and Cmax were 44% and 26% higher in female subjects, respectively, compared to male subjects. The observed exposure difference by sex in healthy subjects is not considered clinically meaningful.Geriatric Patients
Following a single oral dose of 100 mg lonafarnib in healthy subjects, the plasma lonafarnib exposure AUC and Cmax were 59% and 27% higher in subjects ≥65 years, respectively, compared to subjects 18 to 45 years of age. The observed higher exposure in geriatric subjects is not considered clinically relevant.Drug Interaction Studies
In Vitro Studies
Lonafarnib is a CYP3A substrate and a potent CYP3A time-dependent and mechanism-based inhibitor. Lonafarnib is an inhibitor of CYP2C8 and CYP2C19. Lonafarnib is not considered an inhibitor of CYP1A2, CYP2B6, CYP2C8, CYP2C9, or CYP2D6. Lonafarnib is unlikely to be an inducer of CYP1A2, CYP2B6, and CYP3A.Lonafarnib is not a substrate of transporters OATP1B1, OATP1B3, or BCRP, but is likely a marginal substrate of P-gp. Lonafarnib is an inhibitor of P-gp, OATP1B1, OATP1B3, and BCRP.
Clinical Studies: Effects of other Drugs on Lonafarnib
CYP3A inhibitors
Lonafarnib is a sensitive substrate for CYP3A. With coadministration of a single oral dose of 50 mg lonafarnib following 200 mg ketoconazole (a strong CYP3A inhibitor) once daily for 5 days, the Cmax and AUC of lonafarnib were increased by 270% and 425%, respectively, as compared to lonafarnib administered alone in healthy subjects [see Dosage and Administration (2.3), Contraindications (4), Drug Interactions (7.1)].CYP2C9 inhibitors
Coadministration with CYP2C9 inhibitors may increase lonafarnib AUC and Cmax. A drug-drug interaction study of ZOKINVY with CYP2C9 inhibitors has not been conducted [see Drug Interactions (7.1)].CYP3A inducers
With coadministration of a single oral dose of 50 mg lonafarnib (combined with a single oral dose of 100 mg ritonavir) following 600 mg rifampin once daily for 8 days, the Cmax of lonafarnib was reduced by 92% and the AUC was reduced by 98%, as compared to without rifampin coadministration in healthy subjects [see Contraindications (4), Drug Interactions (7.1)].Clinical Studies: Effects of Lonafarnib on other Drugs
CYP3A Substrates
Lonafarnib is a strong inhibitor of CYP3A. With coadministration of a single oral dose of 3 mg midazolam with multiple oral doses of 100 mg lonafarnib twice daily for 5 days in healthy subjects, the Cmax and AUC of midazolam were increased by 180% and 639%, respectively [see Dosage and Administration (2.4), Contraindications (4), Drug Interactions (7.2)].Loperamide
With coadministration of a single oral 2 mg dose of loperamide (primarily metabolized by CYP2C8 and CYP3A and a substrate of P-gp) with multiple oral doses of lonafarnib 100 mg twice daily for 5 days in healthy subjects, the Cmax and AUC of loperamide were increased by 214% and 299%, respectively [see Drug Interactions (7.2)].CYP2C19 Substrates
Lonafarnib is a moderate CYP2C19 inhibitor. With coadministration of a single oral dose of 40 mg omeprazole with multiple oral doses of lonafarnib 75 mg twice daily for 5 days in healthy subjects, the Cmax and AUC of omeprazole were increased by 28% and 60%, respectively [see Drug Interactions (7.2)].P-gp and OATP1B Substrates
With coadministration of a single oral dose of 180 mg fexofenadine (a P-gp and OATP1B substrate) with multiple oral doses of 100 mg lonafarnib twice daily for 5 days in healthy subjects, the Cmax and AUC of fexofenadine were increased by 21% and 24%, respectively [see Drug Interactions (7.2)].13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Carcinogenicity studies have not been conducted with lonafarnib.Mutagenesis
Lonafarnib was not genotoxic in the bacterial mutagenicity (Ames) assay, in vitro chromosomal aberration assay in mammalian cells, or in vivo micronucleus assay in mice.Impairment of Fertility
Lonafarnib produced impaired fertility in male rats at 90 mg/kg/day or higher (1.5 times the AUC in humans at the recommended dose of 150 mg/m2 twice daily), with a nearly complete loss of fertility at 180 mg/kg/day (3 times the AUC in humans). Male rats treated with 180 mg/kg/day exhibited small testes, flaccid testes, and discolored epididymis (84%, 56%, and 24% of males, respectively). No effects on fertility occurred in males at systemic exposures lower than the human AUC at 150 mg/m2 twice daily.Female rats treated with 30 mg/kg/day lonafarnib or higher (1.2 times the human AUC at the recommended human dose of 150 mg/m2 twice daily) showed a decrease in fertility, as indicated by reductions in the number of corpora lutea and implantation sites, and increases in pre- and post-implantation loss. No effects on fertility occurred in females at systemic exposures lower than the human AUC at 150 mg/m2 twice daily [see Warnings and Precautions (5.5)].
13.2 Animal Toxicology and/or Pharmacology
Renal toxicity occurred in a 6-month oral toxicity study of lonafarnib in rats, with kidney lesions (interstitial necrosis and mineralization in the inner medulla) and correlating changes in clinical chemistry (e.g., hyperphosphatemia, hyperkalemia) and urinalysis parameters observed at systemic exposures approximately equal to the AUC in humans at the recommended dose of 150 mg/m2 twice daily. No evidence of renal toxicity was observed at systemic exposures lower than the human AUC at 150 mg/m2 twice daily [see Warnings and Precautions (5.3)].
Toxicity in the male reproductive tract occurred in a 1-year oral toxicity study in monkeys at 10 mg/kg/day or higher (AUC lower than the human AUC at the recommended dose of 150 mg/m2 twice daily). The lesions in the male reproductive tract included aspermia in the epididymis and atrophy of seminiferous tubules, seminal vesicle, and prostate gland. Testicular toxicity was also observed in rats, where severe impairment of male fertility occurred [see Warnings and Precautions (5.5), Nonclinical Toxicology (13.1)].
Ocular (retinal) toxicity occurred in a 1-year oral toxicity study in monkeys at 40 mg/kg/day (3.7 times the human AUC at the recommended dose of 150 mg/m2 twice daily). The retinal injury involved single cell necrosis of photoreceptor cells in the layer of rods and cones and the outer nuclear layer. No retinal toxicity was observed at 20 mg/kg/day (2.1 times the human AUC at 150 mg/m2 twice daily). However, in a follow-up study of lonafarnib effects on visual function in monkeys as evaluated by electroretinography, oral administration of 15 mg/kg/day for 13 weeks or 60 mg/kg/day for 6 weeks produced adverse effects on rod-dependent, low-light vision. The effects were observed at several time-points throughout the treatment period. No histological changes in the retina were observed at study termination [see Warnings and Precautions (5.4)].
14 CLINICAL STUDIES
The efficacy of ZOKINVY is based on results from the Observational Cohort Survival Study, which retrospectively compared survival data from two Phase 2 studies in patients with HGPS to those from a natural history cohort.
Study 1 (NCT00425607) was a Phase 2 open-label, single-arm trial that evaluated the efficacy of ZOKINVY in 28 patients (26 with classic HGPS, one with non-classic HGPS, and one with Progeroid Laminopathy with LMNA heterozygous mutation with progerin-like protein accumulation). Patients received ZOKINVY for 24 to 30 months. Patients initiated treatment with ZOKINVY 115 mg/m2 twice daily. After 4 months of treatment, patients who tolerated treatment had an increase in dose to 150 mg/m2 twice daily. Among the 28 patients treated, 27 patients with HGPS (16 females, 11 males) were included in the survival assessment. The median age at treatment initiation for the 27 patients was 7.5 years (range: 3 to 16 years). The body weight range was 6.6 to 17.6 kg and the BSA range was 0.38 to 0.75 m2 (ZOKINVY is not indicated in patients with a BSA less than 0.39 m2 because the appropriate dosage strength is not available for this population).
Following completion of Study 1, 26 patients enrolled in a second Phase 2 open label, single-arm trial (Study 2, NCT00916747) which consisted of two study phases. In the first phase of Study 2, patients received ZOKINVY with additional therapies for about 5 years. In the second phase of Study 2, patients received ZOKINVY 150 mg/m2 twice daily for a period of up to 3 years.
There were 35 treatment naïve patients with HGPS enrolled into the second phase of Study 2. Among the 35 treated patients (22 males, 13 females), 34 (97.1%) patients had classic HGPS and 1 (2.9%) patient had non-classic HGPS. The median age was 6 years (range: 2 to 17 years). The body weight range was 6.7 to 22 kg and the BSA range was 0.42 to 0.90 m2.
Throughout Study 1 and Study 2, ZOKINVY was administered orally via capsules or the capsule contents were mixed with Ora Blend SF or Ora-Plus and administered orally as a suspension.
The retrospective survival analysis was based on the mortality data from 62 treated patients (27 patients in Study 1 and 35 treatment-naïve patients in Study 2) and data from matched, untreated patients in a separate natural history cohort. The lifespan of HGPS patients treated with ZOKINVY increased by an average of 3 months through the first three years of follow-up and 2.5 years through the last follow-up time (11 years) compared to untreated patients. The survival analysis summary is provided in Table 7 and Figure 1.Table 7: Survival Analysis Summary for Patients with HGPS

Figure 1: Kaplan-Meier Survival Curves for Follow-up Time Censored at Last Follow-up for Patients with HGPS
Note: The Kaplan-Meier (KM) survival curve for the ZOKINVY-treated patients is indicated with a solid line; the curve for the untreated patients is indicated with a dashed line. The shaded regions in blue and red represent the 95% confidence bands for the treated and untreated KM survival curves, respectively.
16 HOW SUPPLIED/STORAGE AND HANDLING
ZOKINVY is supplied as:
- 50 mg capsules: Size 4 hard capsule, opaque yellow with “LNF” and “50” printed in black.
Bottles of 30 capsules each (NDC 73079-050-30) - 75 mg capsules: Size 3 hard capsule, opaque light orange with “LNF and “75” printed in black.
Bottles of 30 capsules each (NDC 73079-075-30)
Store at 20°C-25°C (68°F-77°F), excursions permitted to 15°C-30ºC (59°F-86°F) [see USP Controlled Room Temperature].
17 PATIENT COUNSELING INFORMATION
.Advise the patient to read the FDA-approved patient labeling (Patient Information and Instructions for Use).
Dosing [see Dosage and Administration (2.1)]
- Advise patients and caregivers that ZOKINVY should be taken twice daily with the morning and evening meals.
- Inform patients and caregivers that if a dose is missed, the next dose should be given as soon as possible up to 8 hours prior to the next scheduled dose. If less than 8 hours remain before the next scheduled dose, the patient should skip the missed dose and resume taking ZOKINVY at the next scheduled dose
Preparation and Administration [see Dosage and Administration (2.5), Drug Interactions (7)]
- Advise patients to swallow the capsule whole with water. The capsules should not be chewed.
- For patients unable to swallow capsules, advise patients and caregivers that the contents of ZOKINVY can be mixed with Ora Blend SF or Ora-Plus. For patients unable to access or tolerate Ora Blend SF or Ora-Plus, the contents of ZOKINVY can be mixed with orange juice or applesauce. Advise patients not to mix the contents of ZOKINVY with juice containing grapefruit or Seville oranges. Advise patients and caregivers that the mixture must be prepared fresh for each dose and taken within approximately 10 minutes of mixing.
- Advise patients and caregivers to read and carefully follow the instructions for administering the capsule contents in Ora Blend SF, Ora-Plus, orange juice or applesauce [see Instructions for Use]. Advise patient and caregivers to call their healthcare provider or pharmacist if they have any questions.
Drug Interactions [see Dosage and Administration (2.3, 2.4), Contraindications (4), Warnings and Precautions (5.1), Drug Interactions (7)]
Inform patients and caregivers that ZOKINVY may interact with many drugs. Advise patients and their caregivers to report the patient’s use of all prescription and nonprescription medications, including nutritional supplements and vitamins.Gastrointestinal Adverse Reactions [see Dosage and Administration (2.2), Adverse Reactions (6.1)]
Inform patients and caregivers that gastrointestinal adverse reactions are common with ZOKINVY. These include, but are not limited to, vomiting, diarrhea, and nausea. Advise patients and caregivers to contact their healthcare provider if these adverse reactions persist.Hypertension [see Adverse Reactions (6.1)]
Inform patients and caregivers that blood pressure may increase while taking ZOKINVY. Symptoms of hypertension may include headaches, shortness of breath, nosebleeds, flushing, dizziness, or chest pain. Advise patients and caregivers to contact their healthcare provider if these adverse reactions occur.Nephrotoxicity [see Warnings and Precautions (5.3), Nonclinical Toxicology (13.2)]
Inform the patient and caregiver of the risk of kidney damage.Retinal Toxicity [see Warnings and Precautions (5.4), Nonclinical Toxicology (13.2)]
Inform the patient and caregiver of the risk of developing difficulty with night vision. Advise patients and caregivers to contact their healthcare provider if they experience a change in vision.Impaired Fertility [see Warnings and Precautions (5.5), Nonclinical Toxicology (13.1)]
Inform females and males of reproductive potential that ZOKINVY may impact pubertal development and impair fertility.Embryo-Fetal Toxicity [see Warnings and Precautions (5.6), Use in Specific Populations (8.1, 8.3)]
Inform pregnant women and female patients of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with ZOKINVY.Manufactured for:
Eiger BioPharmaceuticals, Inc., 2155 Park Boulevard Palo Alto, CA 94306
ZOKINVY is a trademark of Eiger BioPharmaceuticals, Inc.

PRINCIPAL DISPLAY PANEL - NDC: 73079-050-30 - 50 mg 30-count Bottle Label
PRINCIPAL DISPLAY PANEL - NDC: 73079-050-30 - 50 mg 30-count Bottle Carton Label
PRINCIPAL DISPLAY PANEL - NDC: 73079-075-30 - 75 mg 30-count Bottle Label
PRINCIPAL DISPLAY PANEL - NDC: 73079-075-30 - 75 mg 30-count Bottle Carton Label
【备注】以上内容仅供参考,不作为用药依据,详情请参照药品附带说明书。